Abstract
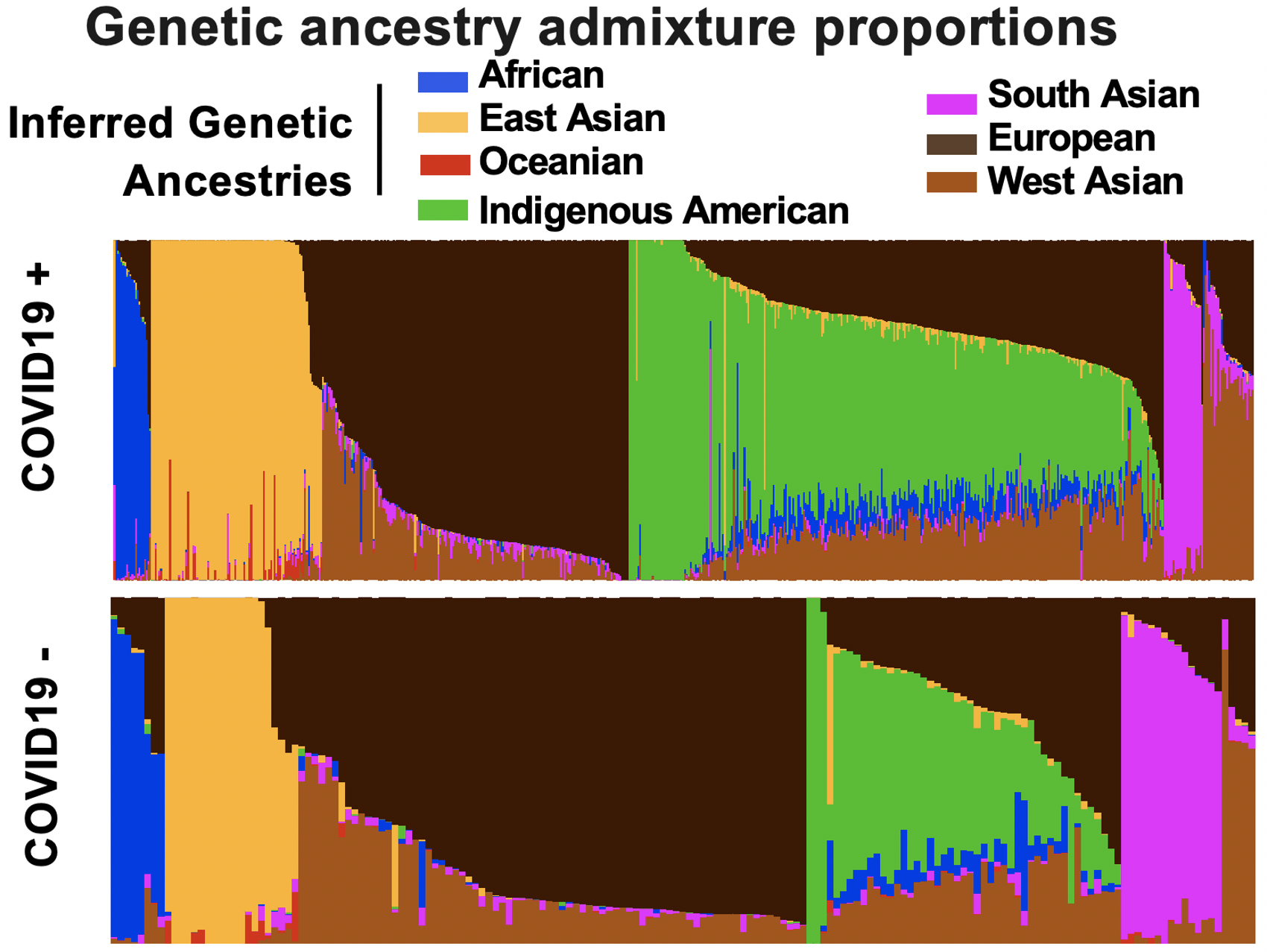
The SARS-CoV-2 pandemic has differentially impacted populations across race and ethnicity. A multi-omic approach represents a powerful tool to examine risk across multi-ancestry genomes. We leverage a pandemic tracking strategy in which we sequence viral and host genomes and transcriptomes from nasopharyngeal swabs of 1049 individuals (736 SARS-CoV-2 positive and 313 SARS-CoV-2 negative) and integrate them with digital phenotypes from electronic health records from a diverse catchment area in Northern California. Genome-wide association disaggregated by admixture mapping reveals novel COVID-19-severity-associated regions containing previously reported markers of neurologic, pulmonary and viral disease susceptibility. Phylodynamic tracking of consensus viral genomes reveals no association with disease severity or inferred ancestry. Summary data from multiomic investigation reveals metagenomic and HLA associations with severe COVID-19. The wealth of data available from residual nasopharyngeal swabs in combination with clinical data abstracted automatically at scale highlights a powerful strategy for pandemic tracking, and reveals distinct epidemiologic, genetic, and biological associations for those at the highest risk.